
Mi chiamo Elena Olmastroni, ho una laurea magistrale in Biostatistica e sono iscritta al primo anno di dottorato in Scienze farmacologiche biomolecolari, sperimentali e cliniche del DiSFeB. Attualmente svolgo la mia attività di ricerca presso il Servizio di epidemiologia e farmacologia preventiva (SEFAP), diretto da Alberico L. Catapano.
La genetica umana e la genetica statistica sono passate da settori relativamente oscuri a quelli tra i più entusiasmanti e discussi della scienza moderna. Questo perché nel tempo l’epidemiologia genetica, che studia i fattori genetici e la loro interazione con i fattori ambientali nella frequenza di malattie, è diventata un utile supporto all’epidemiologia osservazionale, che studia le caratteristiche delle malattie (frequenza, andamento, ecc.) a livello di popolazione, per comprendere gli aspetti ereditari delle patologie e la predisposizione individuale a queste, soprattutto in un’ottica di “cura personalizzata”.
L’incontro tra queste due scienze nasce da un’esigenza ben precisa: ottenere solide evidenze scientifiche osservando/studiando soggetti “reali”, ovvero donne, uomini e anziani che sono sottoposti alla pratica clinico-assistenziale quotidiana, caratterizzati da problemi clinici compositi e/o malattie croniche, che spesso comportano l’assunzione di molti farmaci. In altre parole, ci piacerebbe studiare soggetti che per la loro “complessità” non verrebbero mai reclutati in studi clinici controllati e randomizzati (RCTs), che da sempre sono considerati il gold-standard della ricerca clinica, ovvero il disegno di studio più accurato per confermare un determinato dubbio diagnostico.
Cerchiamo di capire perché questo potrebbe essere un problema. Se ci pensiamo bene il quesito che vogliamo risolvere è teoricamente molto semplice: nella pratica clinica ho dei soggetti che sono esposti ad un trattamento farmacologico o ad una determinata condizione ed altri che non lo sono, e voglio capire se questi due gruppi hanno la stessa probabilità di sperimentare un particolare evento o se hanno la stessa probabilità di beneficiare dell’esposizione a cui sono sottoposti. Il problema è che, in questa semplice relazione, si inseriscono innumerevoli variabili e condizioni (uso di altri farmaci, stile di vita, fattori ambientali, patologie concomitanti) specifiche per ogni individuo, che in qualche modo hanno un impatto, più o meno forte, nell’associazione che vogliamo studiare. Questo significa che, per valutare se esiste un vero rapporto causale tra l’esposizione e l’evento, dovrei cercare di tenere sotto controllo l’effetto “confondente” di tutti questi fattori, ma anche l’effetto di altre componenti che purtroppo non riesco a misurare (ad esempio non sempre è disponibile l’informazione sull’abitudine al fumo degli individui che osservo) ma che potrebbero comunque avere un impatto importante nella valutazione della relazione in esame. Niente di più complesso!
È proprio per questo che ci dispiace allontanarci dal settingdegli RCTs. In questi studi i partecipanti vengono assegnati in maniera casuale (tecnica di randomizzazione) al gruppo di esposti o al gruppo di controllo (soggetti non esposti), e questo permette di distribuire equamente tutti gli altri fattori prognostici tra i due gruppi di pazienti, tra i quali l’unica differenza risulta essere solo l’intervento in studio. Nel contesto osservazionale tutto questo non è possibile: posso solo osservare le differenze negli esiti che si verificano dopo che le decisioni, come effettuare un determinato trattamento, sono già state prese.
E se ci fosse però un modo di ricreare la tecnica di randomizzazione anche nel contesto osservazionale? Immaginate i vantaggi: da un lato poter suddividere la mia popolazione in gruppi omogenei che differiscono solo per il fattore che voglio studiare, dall’altro lato poter studiare questa relazione in un campione molto eterogeneo che mi consente di ottenere risultati generalizzabili all’intera popolazione.
Dovremmo trovare, per stratificare la popolazione, una variabile super partes che:
- sia associata in maniera esclusiva con l’esposizione in esame
- non sia associata con quei fattori definiti “confondenti”
- sia in relazione con l’evento che vogliamo studiare solo tramite l’esposizione (nessuna associazione diretta).
Assumendo una posizione neutrale rispetto a tutti i possibili confondenti che possono interferire, questa variabile potrebbe essere utilizzata per suddividere i soggetti in gruppi, nei quali le condizioni cliniche, anche quelle più complesse, sono equamente distribuite, proprio come avviene tramite la randomizzazione.
In realtà, la soluzione a questo rompicapo è sempre stata davanti ai nostri occhi (o meglio, dentro di noi!), e per questo dobbiamo ringraziare Gregor Johann Mendel, precursore della moderna genetica, e le sue 28.000 piante di piselli odorosi che coltivò e analizzò durante ben sette anni di esperimenti.
Mendel ci ha dimostrato che i geni che ereditiamo dai nostri genitori ci vengono trasferiti in maniera casuale al momento del concepimento (legge della segregazione dei caratteri), e che durante la formazione dei gameti, geni diversi si distribuiscono l’uno indipendentemente dall’altro (legge dell’assortimento indipendente). Questo significa che gli individui di una popolazione ereditano casualmente un certo allele di una data caratteristica (variante genetica), così come i partecipanti di un RCT ricevono casualmente il trattamento che viene testato, e che per definizione questa variante genetica non può essere modificata o influenzata da altri caratteri (almeno in teoria).
Abbiamo trovato la variabile che stiamo cercando: basterà identificare una variante genetica che rispetti i 3 criteri sopra citati ed il gioco è fatto!
Facciamo un esempio pratico. Supponiamo di voler studiare l’associazione tra alti livelli di colesterolo LDL (LDL-C) ed insorgenza di eventi cardiovascolari (CV) in una generica popolazione, ma che sia difficile dare una stima di questa associazione che sia il più possibile precisa, visto che altri fattori potrebbero influire non solo sui livelli di LDL-C, ma anche sulla probabilità di sperimentare un evento CV, come ad esempio elevati livelli di glicemia, lo stato di menopausa nelle donne o l’abitudine al fumo.
Supponiamo però di essere in grado di suddividere questa popolazione in due gruppi, uno composto da soggetti che hanno una forma alternativa di un gene, ad esempio l’allele A associato a livelli medi di LDL-C, e un gruppo di individui che hanno l’allele B per la stessa variante genetica associata però con livelli elevati di LDL-C. In questo modo per suddividere la nostra popolazione non abbiamo usato livelli misurati di LDL-C, che in un determinato istante potrebbero essere influenzati da altri fattori che impattano anche sull’evento, ma abbiamo utilizzato un’informazione genetica, presente nel nostro corredo genetico sin dalla nascita, che indica la predisposizione per ogni individuo ad avere quei livelli di LDL-C osservati. Poiché le caratteristiche genetiche sono distribuite casualmente rispetto ad altri tratti, fattori ambientali e ad altri fattori di rischio nella popolazione, il tasso di insorgenza di eventi CV può essere confrontato direttamente tra i soggetti con allele A e l’allele B. Una differenza nel rischio di malattia tra i due gruppi, individuata tramite la variante genetica (associata solo con i livelli di LDL-C), indicherà direttamente un effetto causale del fattore di rischio (LDL-C) sulla malattia cardiovascolare.
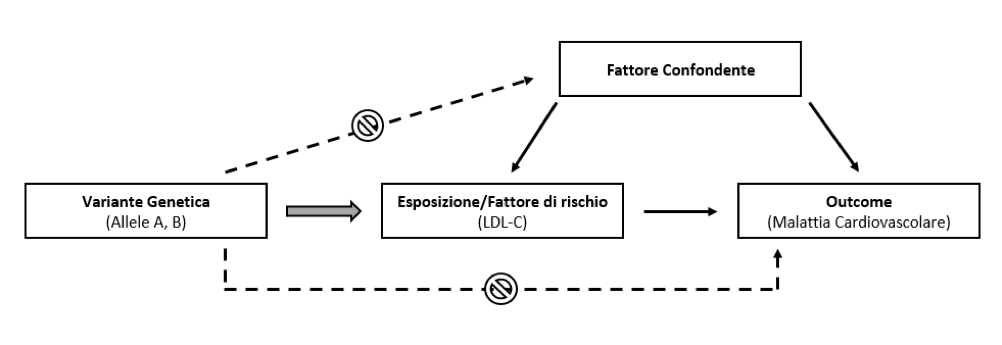
Questo tipo di disegno di studio prende il nome di “Randomizzazione Mendeliana”. Alla base di questa tecnica vi sono assunti metodologici specifici e stringenti che però, se rispettati, forniscono nuove opportunità per testare la causalità, esplorare i meccanismi biologici, e capire/prevenire gli effetti avversi delle esposizioni modificabili (stile di vita, dislipidemia, ipertensione arteriosa, ecc…) sulla salute umana. In questo contesto, le varianti genetiche che vengono utilizzate come strumento per studiare in maniera mediata un’associazione di interesse, vengono definite appunto “variabili strumentali”.
Questa nuova prospettiva ha suscitato molto interesse in diversi ambiti, e in particolare in quello cardiovascolare, che da un paio di anni è diventato il filo conduttore di quasi tutte le mie analisi.
L’identificazione di varianti genetiche associate al rischio di malattia CV e dei rispettivi biomarkers, ovvero frammenti della sequenza di DNA che possono essere responsabili dell’insorgenza della malattia o di una predisposizione patologica, è stata l’obiettivo principale della ricerca genetica CV a partire dai primi studi di associazione genome-wide.
Nel tempo, gli studi di randomizzazione mendeliana hanno permesso da un lato di confermare geneticamente il ruolo predittivo, ovvero la capacità di predire l’insorgenza della malattia, di molti fattori di rischio cardiovascolare, come ad esempio per l’associazione con elevati valori di LDL-C, della Lipoproteina (a), della pressione sanguigna e dell’indice di Massa Corporea (BMI), dall’altro lato invece hanno messo in dubbio il ruolo causale di altri fattori, come il colesterolo HDL, l’associazione con la malattia renale cronica, con i livelli di vitamina C e tanti altri ancora, ruolo che era stato confermato in passato da studi osservazionali e RCTs.
Ad oggi abbiamo a disposizione tante evidenze circa diverse varianti genetiche che giocano un ruolo chiave nella predisposizione alla malattia CV. Tuttavia, sono evidenze che solo se connesse/legate tra di loro, possono darci una stima del rischio CV globale per ogni individuo.
Da qui nasce l’idea del mio progetto: combinare l’informazione proveniente da diverse varianti genetiche associate con la malattia CV in un punteggio di rischio genetico individuale che possa predire il rischio CV globale (ovvero che tenga conto contemporaneamente di tutti i fattori di rischio) per ogni individuo in maniera robusta e validata. E perché non sfruttare le conoscenze biologiche esistenti sul metabolismo del LDL-C? Partendo dalle varianti genetiche individuate potrebbe essere possibile confrontare l’effetto di queste, che agiscono su una specifica proteina o su una porzione della via metabolica, con l’effetto di un farmaco che agisce sullo stesso bersaglio. Questo permetterebbe non solo di verificare l’efficacia di un trattamento a lungo termine, ma anche di anticipare i segnali di sicurezza di alcuni trattamenti CV ed identificare nuovi bersagli terapeutici.
Lascia un commento