
La disabilità intellettiva (ID) è una condizione che si verifica nel periodo antecedente i 18 anni ed è dovuta ad un non corretto sviluppo del cervello. L’ID rappresenta un gruppo eterogeneo di disturbi caratterizzati da un’intelligenza sotto la media, dall’incapacità di svolgere le azioni quotidiane e da un ritardo nell’apprendimento.
Il termine disabilità intellettiva è di recente utilizzo e sostituisce, dal 2013, quello di ritardo mentale, ormai caduto in disuso a causa della sua natura offensiva. La disabilità intellettiva colpisce dall’1 al 3% della popolazione mondiale ed è il disturbo dello sviluppo più comune, rappresentando un importante problema socio-economico nell’assistenza sanitaria.
Le persone con ID presentano deficit principalmente in due aree:
- funzionamento intellettivo, altrimenti noto come QI: si riferisce all’abilità di una persona nel ragionare, prendere decisioni e risolvere problemi. Viene diagnosticata una disabilità intellettiva se il QI è inferiore a 70.
- comportamenti adattativi: sono le capacità necessarie per la vita quotidiana, come il comunicare in modo efficace, interagire con la gli altri e prendersi cura di sé. Viene valutato come il bambino mangia, si veste, come si interfaccia con la famiglia, gli amici e altri bambini.
Esistono quattro livelli di disabilità intellettiva, da lieve a severa, in base al QI. Nelle disabilità gravi possono insorgere problemi più rilevanti come convulsioni, autismo, difficoltà motorie, problemi visivi e uditivi.
Le cause che portano alla disabilità intellettiva sono molteplici e sono riassunte nella tabella sottostante.

L‘eterogeneità della disabilità intellettiva ne rende difficile la diagnosi genetica e clinica e molte varianti sono ancora difficili da interpretare.
Tuttavia, un approccio combinato di sequenziamento del genoma su larga scala e analisi funzionale, elettrofisiologica e bioinformatica si sta dimostrando molto efficace per comprendere le cause dell’ID e aiutare ad interpretare i nuovi geni che la causano.
Una ricerca attraverso i database OMIM e SysID rivela che circa 400 geni diversi possono causare ID autosomica recessiva. L’ID autosomica dominante, invece, causata da mutazioni in eterozigosi, conta attualmente circa 180 geni. Infine, circa il 10-12% dei geni del cromosoma X umano è stato collegato con la disabilità intellettiva.
Tuttavia, anche se i geni identificati sono molteplici, bisogna ricordare che i due terzi dei casi di disabilità intellettiva sono a causa ignota e i meccanismi molecolari alla base della patologia rimangono sconosciuti. Per questo motivo diventa importante studiare i meccanismi molecolari attraverso modelli sperimentali.
Una collaborazione con il Fernández-Jaén dell’Hospital Universitario Quirónsalud di Madrid ci ha permesso di individuare un paziente affetto da una disabilità intellettiva grave, combinando il sequenziamento dell’esoma con analisi in silico. Il paziente presenta una mutazione de novo in eterozigosi nel gene SEMA3E ed in particolare si ha la delezione di una base che porta ad un frameshift, generando un codone di stop prematuro e la produzione di una proteina tronca.
Il gene SEMA3E codifica per la semaforina 3E, una proteina chiave nel controllo dello sviluppo cerebrale murino. Le semaforine, in generale, sono molecole chiave nel mediare la comunicazione cellula-cellula e nel controllare una varietà di funzioni cellulari.
Le mutazioni delle semaforine e dei loro recettori, neuropiline e plexine, sono state finora collegate principalmente alla mancanza di ormone di rilascio delle gonadotropine (GnRH). Studi precedenti del nostro laboratorio, per esempio, hanno individuato una correlazione tra una mutazione in eterozigosi della semaforina 3e con difetti a livelli dei neuroni secernenti GnRH. Tuttavia, recenti evidenze supportano fortemente che questa classe di molecole possa anche essere implicata nelle malattie del neurosviluppo, come per esempio i disturbi dello spettro autistico.
Attraverso dei saggi in vitro ed ex vivo, nel laboratorio Cariboni abbiamo dimostrato che la mutazione porta ad una perdita di funzione del gene SEMA3E, che fa sì che la proteina non venga secreta e di conseguenza non si leghi al suo recettore plexina D1, sia in cellule neuronale murine che in tessuti murini e umani.
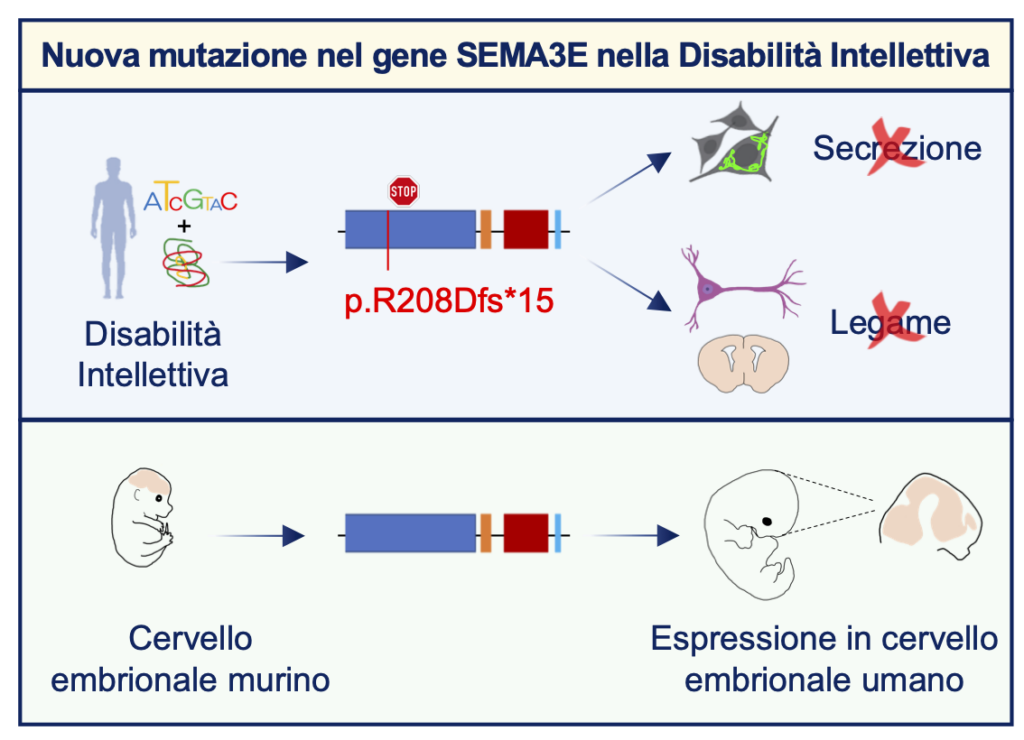
Lo studio in modelli sperimentale knokout per la Sema3e ha evidenziato dei difetti dello sviluppo neurologico, altamente correlati ai segni clinici del nostro paziente, che includono regressione cognitiva, disabilità intellettiva, compromissione della memoria e tic. È quindi possibile ipotizzare che le caratteristiche neurologiche cliniche del paziente possano essere dovute, almeno in parte, ad un signalling difettoso della SEMA3E durante lo sviluppo embrionale del cervello.
Saranno necessari ulteriori studi per comprendere il ruolo delle semaforine nei disturbi del neurosviluppo e scoprire un nuovo tassello in una sintomatologia per lo più idiopatica.
Lascia un commento