
La sindrome CHARGE (CS), descritta per la prima volta nel 1979, è una malattia genetica rara che colpisce circa uno su 15.000 nuovi nati. Si tratta di una patologia causata da difetti dello sviluppo embrionale a carico di diversi organi quali il cuore, il cervello, i nervi cranici, gli occhi, le orecchie e l’apparato genitale. Il termine CHARGE è un acronimo inglese delle più comuni manifestazioni cliniche con cui si manifesta la sindrome: Coloboma dell’occhio, difetti cardiaci (Heart), Atresia delle coane nasali, Ritardo di crescita e dello sviluppo cognitivo, anomalie a carico dei Genitali e/o urinarie, anomalie dell’orecchio (Ear) interno ed esterno. Sono stati inoltre osservati casi di pazienti con sintomi neurologici specifici, tra cui disturbi dello spettro dell’autismo ed epilessia.
Il tasso di mortalità associato a CS nei primi cinque anni di vita è circa del 30% e l’aspettativa di vita dipende largamente dalla gravità dei sintomi. Nel 2004 è stato scoperto il gene responsabile di questa malattia: il gene Chromodomain Helicase DNA binding protein 7 (CHD7), che codifica per una proteina capace di modificare la struttura del DNA e regolare lo sviluppo fetale di diversi organi e tessuti. Tra questi vi è anche il Sistema Nervoso Centrale (SNC), dove CHD7 controlla la proliferazione e differenziazione dei progenitori neurali.
La trasmissione di questa malattia genetica è di tipo autosomico dominante. Pertanto, una sola copia difettosa del gene è sufficiente per manifestare i sintomi della malattia. Il 60-70% dei pazienti presenta mutazioni de novo, cioè sporadiche, mentre solo raramente è stata documentata una trasmissione di tipo familiare.

CHD7 fa parte di una famiglia di proteine coinvolte nella regolazione dello stato della cromatina. Questi fattori epigenetici sono in grado di utilizzare l’energia dell’ATP per muoversi sul DNA, rimodellando la struttura della cromatina e controllando così l’espressione genica. Questo processo consente ai fattori di trascrizione e al macchinario replicativo di accedere al DNA, preferenzialmente nelle regioni in cui la cromatina è meno condensata.
Ad oggi non è disponibile alcun trattamento farmacologico per la CS. Le uniche opzioni per i bambini affetti risiedono in interventi chirurgici invasivi e visite specialistiche fin dai primi mesi di vita, per correggere alcune delle anomalie congenite. Subito dopo la nascita è infatti fondamentale stabilizzare la respirazione e, quando necessario, utilizzare un’alimentazione tramite sondino nasogastrico. All’esordio dell’età puberale diventa invece necessario procedere con una terapia ormonale sostitutiva per uno sviluppo fisiologico degli organi sessuali. I bambini necessitano inoltre di valutazioni psicologiche frequenti e supporto scolastico. Tutte queste pratiche non sono in grado di curare la sindrome, ma si propongono di alleviare alcuni dei sintomi.
Nonostante oltre l’85% dei pazienti con CS presenti mutazioni in CHD7, il numero dei sintomi manifestati e il loro grado di gravità è estremamente variabile, persino tra individui all’interno di una stessa famiglia. Una possibile spiegazione di questa eterogeneità fenotipica è che mutazioni in altri geni, aventi una relazione epistatica con CHD7, possano alterare l’espressività dei tratti della malattia. In questo contesto, i geni che codificano per le semaforine (SEMA) e i loro recettori sono recentemente emersi quali buoni candidati in grado di interagire con CHD7 nel SNC.
Le SEMA giocano un ruolo centrale nello sviluppo del SNC, orchestrando movimenti cellulari e connessioni che plasmano la sua intricata architettura. Si tratta di proteine che agiscono come segnali guida, dirigendo la migrazione neuronale e la crescita di assoni e dendriti. Attraverso un complesso gioco di attrazione e repulsione, le SEMA scolpiscono dunque il “paesaggio neurale”, garantendo che i neuroni raggiungano le loro destinazioni finali e stabiliscano precise connessioni sinaptiche. La comprensione di questo intricato sistema di segnalazione è fondamentale per studiare i meccanismi sottostanti alla plasticità neuronale e alle disfunzioni neurologiche.
Il laboratorio di Neurobiologia dello Sviluppo coordinato da Anna Cariboni, dove sto svolgendo il mio dottorato di ricerca, si occupa da anni di studiare il ruolo delle SEMA nell’ipogonadismo ipogonadotropo e nei disturbi del neurosviluppo. Il mio progetto di ricerca si propone dunque di sfruttare l’attività tessuto-specifica di CHD7 nel controllo dell’espressione genica per identificare farmaci capaci di migliorare alcuni fenotipi neurologici associati a CS. La nostra idea si basa su aggirare l’assenza di CHD7 e modulare geni bersaglio quali le SEMA e i loro recettori.
A tal proposito, abbiamo messo a punto una pipeline che combina l’uso di due modelli di CS: un ceppo chd-7 mutante del nematode Caenorhabditis elegans (C. elegans) e cellule murine neuronali Chd7-knock-out (Chd7-KO) generate con la tecnica di CRISPR/Cas9.
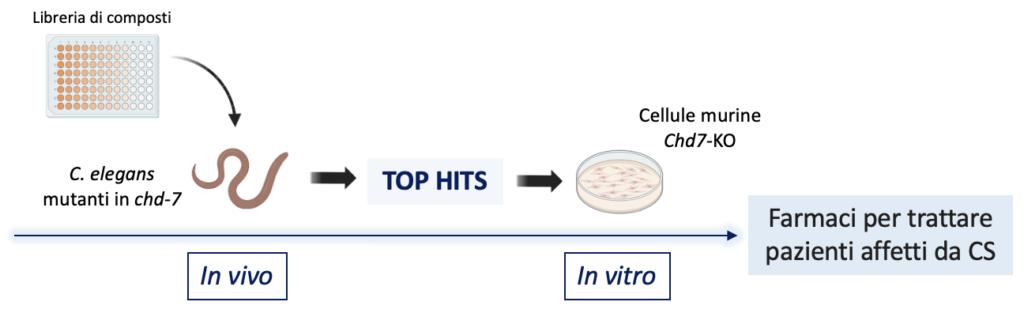
Una vasta libreria di composti che agiscono modulando fattori epigenetici è stata testata sul modello di malattia in C. elegans, ed ha permesso di identificare, in via preliminare, un numero di molecole capaci di migliorare il fenotipo riproduttivo dei mutanti in chd-7. Al contempo, saggi cellulari condotti sulle linee Chd7-KO hanno dimostrato un’importante alterazione della migrazione e proliferazione cellulare. Inoltre, un’analisi trascrittomica mediante RNA Sequencing ha evidenziato alterazioni nei livelli di alcune SEMA. Pertanto, il passo successivo sarà quello di validare le molecole più promettenti nel modello neuronale di CS, valutandone gli effetti sulle alterazioni genetiche e fenotipiche osservate.
Questi esperimenti ci permetteranno di comprendere se sia possibile un trattamento farmacologico in grado di migliorare il fenotipo di malattia mediante una modulazione dei livelli delle SEMA. I risultati di questi studi potranno inoltre essere immediatamente tradotti in altri modelli, e saranno fondamentali per poter pianificare future terapie volte a bypassare l’assenza di CHD7 nei pazienti affetti da CS.
Lascia un commento